Defying Paradigms: A Curiosity-Driven Exploration of pTyr-SNVs in Cancer
Curiosity killed the cat—so the saying goes. But like many scientists, I prefer the extended version: "Curiosity killed the cat, but satisfaction brought it back." It captures how curiosity, though risky, can lead to remarkable discoveries, especially in research.
In science, curiosity is both a blessing and a gamble. Letting it loose can lead to innovative projects but often means deviating from the beaten path your PhD or postdoc advisor laid out for you. This is daunting—venturing into uncharted waters without a guidebook. Yet, if you're fortunate, your curiosity can help you carve out a unique niche and maybe, just maybe, define a new field of research.
For me, curiosity became the engine driving several research projects that now fuel a growing pipeline of manuscripts. I hope one day, this curiosity-driven work will inspire the creation of a new avenue in immuno-oncology. I’d like to share a particular story—how a simple question about a well-studied receptor led me down a path that not only defied established paradigms but opened doors to new ways of understanding cancer progression and anti-tumor immune response.
How It All Began: A Curious Question
In every researcher’s journey, there’s a peak moment when curiosity ignites. For me, that moment came during my postdoc. I had the rare privilege of being in a lab with literally unlimited resources but also complete intellectual freedom. This was key—freedom and independence are essential for curiosity to flourish.
In 2010, I joined Axel Ullrich's lab at the Max Planck Institute of Biochemistry as his last postdoc. Under his mentorship, I had the freedom to apply my expertise in signaling within T cell biology and explore new avenues in signaling biology. This opportunity fueled my long-standing passion for linking membrane receptors to specific transcription factors (TFs). My PhD research focused on T cell-driven autoimmunity, particularly addressing the question of how auto-antigen specific T cells shift from activation to migratory modes. Antigen-specific T cell receptors (TCRs) are notable for several unique features. Despite lacking kinase domains, these receptors act as some of the most potent phosphotyrosine signaling molecules, playing a key role in imparting different differentiation states with distinct functional properties, which ultimately culminates in defending against intracellular pathogens and cancer. One specific aspect of TCR signaling that fascinates me to this day is its ability to recruit cytoplasmic kinases to the inner cell membrane via phosphotyrosine signaling motifs. In fact, this mechanism is effectively exploited by Chimeric Antigen Receptor (CAR) T cell immunotherapies for cancer. When I was introduced to the fibroblast growth factor receptor 4 (FGFR4) project—a problem the lab had been working on for over a decade—it intrigued me.
The FGFR4 p.Gly388Arg variant had been cloned from a cancer cell line as part of a large departmental effort to clone all human kinases. However, despite years of effort, no one could pinpoint a distinct molecular function for this variant. Every hypothesis, focused on the receptor's tyrosine kinase activity, led to dead ends. But instead of being discouraged by the repeated failures, my curiosity was piqued. Why had every avenue failed? What was being overlooked?
A Provocative Hypothesis
The prevailing belief in receptor tyrosine kinase (RTK) biology was that mutations in the extracellular ligand binding domain, transmembrane oligomerization segment, or intracellular kinase domains influence the receptor's activity. However, FGFR4's kinase activity seemed unchanged by the p.Gly388Arg variant, and all downstream MAPK signaling cascades showed no significant differences.
Yet, as a T cell biologist, I couldn’t shake the idea that maybe we were missing something proximal. I proposed a provocative hypothesis: could the variant create a phosphotyrosine motif in the juxtamembrane (jm) region that binds TFs in a novel way, influencing signaling independently of the kinase domain? It was a risky idea, one that defied conventional thinking about RTK signaling—but curiosity demanded I pursue it.
Curiosity Leads to Discovery
Though RTK jm segments are known to undergo autophosphorylation, the dogma held that their role was limited to regulating intrinsic kinase activity. No one had ever considered that a mutation in this region could create a non-canonical binding site for transcription factors, contributing to an entirely different signaling cascade.
To test this hypothesis, I gathered several lines of experimental evidence. All the evidence pointed to a specific elevation of tyrosine 705 phosphorylated STAT3 through membrane-proximal recruitment via the FGFR4 p.Gly388Arg variant. These results were astounding. We discovered that the pTyr site in the germline variant rs351855-G/A, FGFR4 p.Gly388Arg, served as a binding site for STAT3, a transcription factor with important roles in both tumor tissues and the immune system. This unexpected mechanism not only explained the variant's unique signaling but also opened up an entirely new perspective on how germline variations in non-catalytic regions of RTKs could affect both tumor growth and the immune response to the growing tumor.
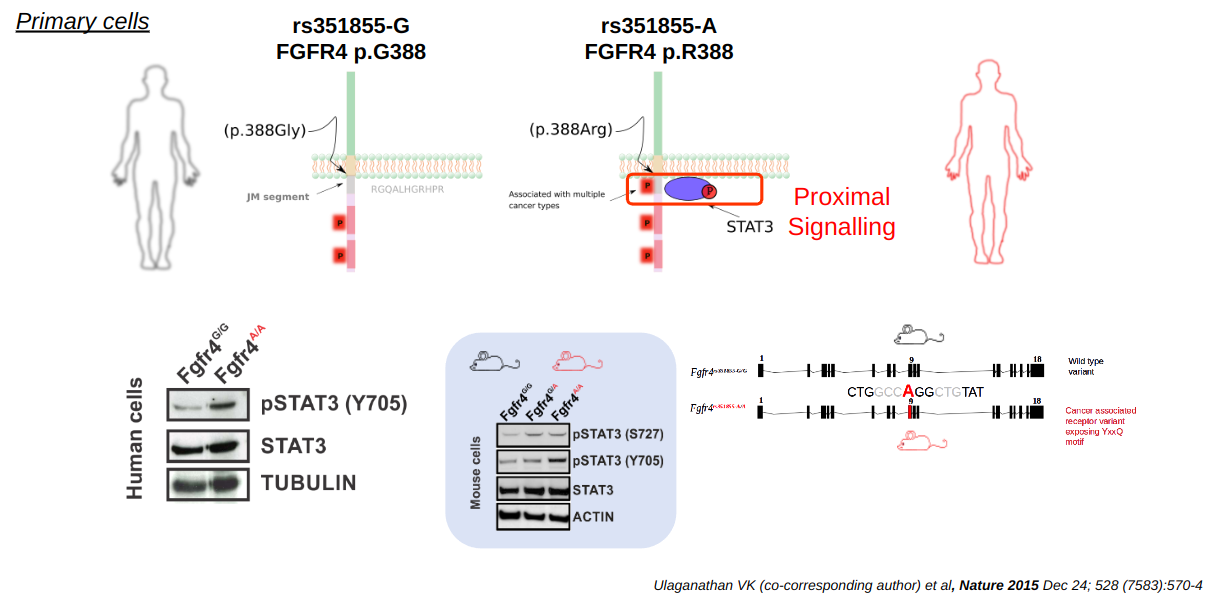
The work was met with both skepticism and excitement. Two out of four reviewers praised the originality of the concept, and we eventually published the findings in Nature (See MPG Press Release). This discovery also laid the foundation for exploring phosphotyrosine-modifying single nucleotide variants (pTyr-SNVs) and their implications in cancer progression, anti-tumor immune response and therapeutic outcomes. Furthermore, we were the first to establish the knowledge that FGFR4 aka CD334 is also expressed highly in regulatory T cells, and that the genetic variant FGFR4 p.Gly388Arg in these cells enhances their suppressive function in the lymph nodes. This led to a homeostasis of lower CD8/Treg ratios and, ultimately, a reduced infiltration of CD8 T cells in the tumor microenvironment (See OMIM *134935).
Intrigued by these findings, I became curious about how many such pTyr-SNVs that create or disrupt proximal signaling exist in the human variome, how many unique non-canonical molecular signaling pathways are waiting to be discovered, and how many are associated with various tumor immune contextures, cancer progression, or therapy outcomes. Answering these questions could take years but has the potential to uncover innovative opportunities for cancer biomarker testing and personalized cancer immunotherapies.
Looking Ahead: Curiosity as a Driving Force in Research
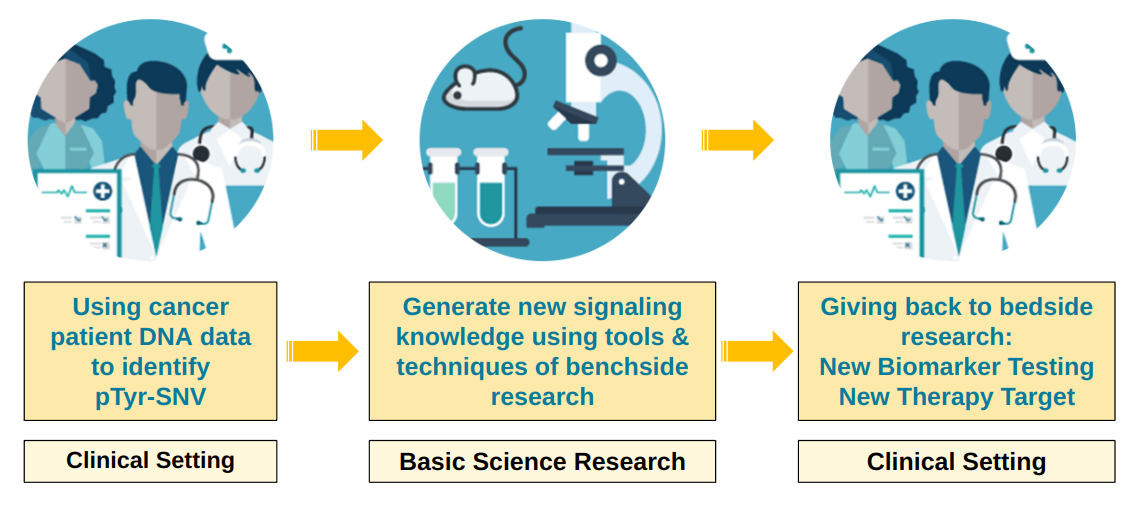
Today, this curiosity-driven work is shaping my research into the role of pTyr-SNVs in immuno-oncology and personalized medicine. By challenging established paradigms and exploring the molecular nuances of germline variants, we are uncovering new biological insights that could significantly impact cancer treatment strategies within a bed-bench-bed research framework (see Figure below). Reflecting on this journey, I realize that the moments where I veered off the expected path—where curiosity led me—were the moments where I made the most progress. For researchers in cancer biology, I offer this: don’t be afraid to follow your questions, even when they defy conventional wisdom. Sometimes, it's the unasked questions, the curious tangents, that lead to the most profound discoveries.
Comments
Post a Comment